22q11.2 Deletion Syndrome (22q11.2 DS) is a rare genetic disease caused by a deletion on chromosome 22 at locus q11.2. 22q11.2 Deletion Syndrome diagnosis is aided by both genetic testing and ultrasound. Accurate antenatal diagnosis is key to appropriate antenatal and neonatal management and can greatly affect 22q deletion life expectancy and quality of life.
22q11.2 DS has also been called DiGeorge syndrome or velocardiofacial syndrome. Today, clinicians recognize that 22q11.2 DS is responsible for these syndromes as well as conotruncal anomaly face syndrome, Opitz G/BBB syndrome, and Cayler cardiofacial syndrome or asymmetric crying facies syndrome. These various syndromes were described before the underlying genetic cause was identified. The various syndromes represent the range of phenotypic expression that can occur with 22q11.2 DS.
Incidence and Genetics
About 90% of cases of 22q11.2 DS are the result of a de novo mutation, although an autosomal dominant inheritance pattern can occur. This syndrome occurs in approximately 1 in 4000 live births and is equally common regardless of sex. At least 36 genes can be affected at this locus, with variability among patients, resulting in a variety of presentations and potential impairments that range from anatomic abnormalities affecting development to cognition and emotional disorders. With proper diagnosis and treatment, life expectancy can be normal; however, the infant mortality of the disease is around 4%, and life expectancy for many is only about 40 years of age. The daily activities of affected individuals may also be severely curtailed.
Abnormalities Associated With 22q11.2 DS
Between 40% and 75% of affected individuals have congenital heart disease, including an interrupted aortic arch, persistent truncus arteriosus, tetralogy of Fallot or a ventricular septal defect. 22q11.2 DS is the most common syndrome associated with these conotruncal disorders. Cyanosis may be present at birth due to heart abnormalities.
Half of affected individuals have a palatal abnormality, including velopharyngeal incompetence (a neuromuscular concern that leads to issues closing the palate), cleft palate and hypertelorism (widely spaced eyes). Feeding problems are common in individuals with palatal abnormalities.
Nearly half of individuals with 22q11.2 DS will have hypoparathryoidism, resulting in hypocalcemia, which can cause seizures. Reduced T-cell numbers may occur due to an absent or diminished thymus, resulting in increased numbers of infections. More than 90% of affected individuals have learning disabilities and as many as 30% of adults develop schizophrenia.
More than 90% of affected individuals have growth or developmental delays, due in part to growth hormone deficiency. Intrauterine growth restriction is also common. Roughly 90% of individuals likewise develop various skeletal abnormalities including club foot, rib anomalies, vertebral differences, polydactyly, and scoliosis. Renal abnormalities and hearing loss can also occur.
Features Observable on Ultrasound
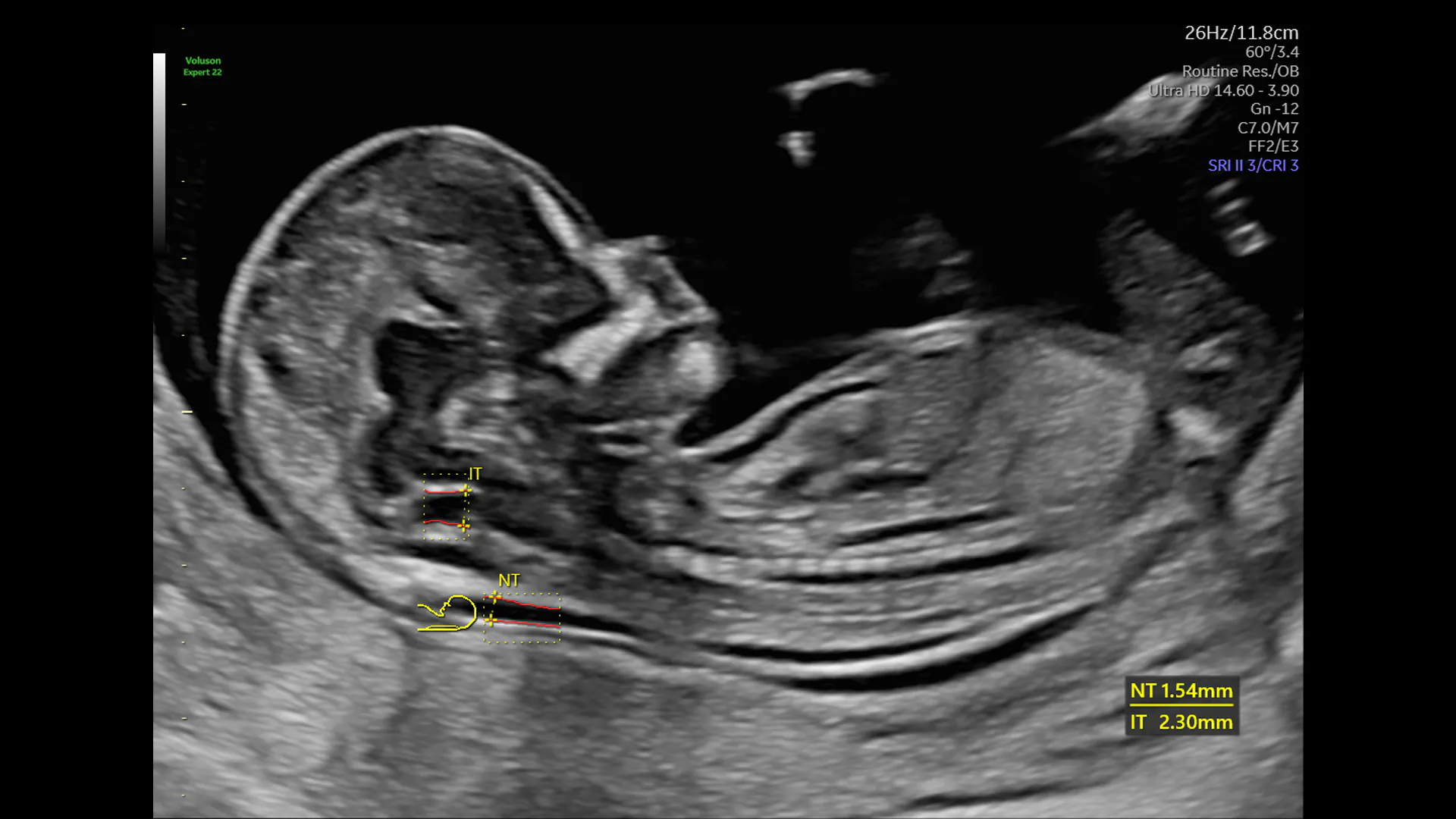
The main features of 22q11.2 DS detectable on ultrasound are cardiac abnormalities, including tetralogy of Fallot, interrupted or right aortic arch, truncus arteriosus, absent pulmonary valve or ventricular septal defect. Any of these findings should prompt consideration of 22q11.2 DS as the possible cause. Other ultrasound findings associated with 22q11.2 DS include unexplained polyhydramnios, thickened nuchal translucency, holoprosencephaly, spina bifida, vertebral anomalies, or renal anomalies. Traditional 2D ultrasound or 3D ultrasound can aid in the detection of hypertelorism and cleft palate, which are common facial findings in 22q11.2 DS. Any combination of these findings on ultrasound should also prompt consideration of 22q11.2 DS and consideration of genetic testing. The incidence of 22q11.2 DS among fetuses with congenital heart disease and an otherwise normal karyotype ranges from 10.8% to as high as 20.7%.
Absence of the thalamus, which can be observed after 15-16 weeks, has a sensitivity of 90% for 22q11.2 DS. Unexplained, early symmetric growth restriction should also prompt consideration of 22q11.2 DS.
Screening and Diagnosis
Non-invasive prenatal tests (NIPTs) using cell-free DNA analysis are currently available for detecting 22q11.2 DS. This technology is currently used widely to screen for trisomy 21 as well as trisomies 13 and 18, though the incidence at birth of 22q11.2 DS is higher than for trisomies 21 and 13 combined. This testing can be used as early as 10 weeks' gestation for general screening or selectively after ultrasound has detected either congenital heart disease or some other constellation of features associated with 22q11.2 DS. Current iterations of this technology have a reported sensitivity of 69.6% and a specificity of 100% for the detection of 22q11.2 DS.
If used as a primary screening test early in pregnancy, the false discovery rate of NIPTs for 22q11.2 DS is approximately 81%. Positive test results are rare, but when a positive result does occur, confirmatory diagnostic testing should be employed — either chorionic villus sampling or amniocentesis, depending on gestational age. NIPTs may also find utility after an anatomic abnormality has been detected on ultrasound; this would change the false discovery rate of the test, since the pre-test probability for 22q11.2 DS would be higher given the ultrasound-detected abnormality. For example, if a conotruncal cardiac abnormality has been discovered with ultrasound, assuming a pre-test probability of 15% (the average of the two previously cited studies), then the false discovery rate drops to just 5%.
Combining ultrasound findings with NIPTs results, whether positive or negative, can give patients a refined risk assessment for fetal 22q11.2 DS. Then, the patient can make a decision about a definitive diagnosis through genetic amniocentesis or chorionic villus sampling before 15 weeks' gestation.
Pregnancy Management After Diagnosis
Once a diagnosis has been established, ultrasound can play a part in regularly surveying for growth restriction as well as polyhydramnios, which is associated with increased difficulty feeding after birth. Detailed and repeated attempts at detecting associated anatomic abnormalities with ultrasound can help prepare for neonatal management. The more that is known about the fetus's anatomic abnormalities prior to delivery by ultrasound detection, the better the neonatal team will be able to address the newborn's immediate needs and necessary assessments. This often includes a prenatal consultation with appropriate subspecialists who can help plan and coordinate care as well as prepare the parents through education and expectation setting.
Pregnancies with a confirmed 22q11.2 deletion syndrome diagnosis should be delivered at a tertiary facility with pediatric cardiology specialists available and neonatal intensive care to treat immediate life-threatening conditions, such as seizures associated with hypocalcemia.
Both ultrasound and cell-free DNA serve an important role in screening for 22q11.2 DS as well as optimizing neonatal outcomes and providing appropriate care to the newborn — in turn minimizing negative outcomes and maximizing life expectancy and quality of life for patients and their families.